-
Documentation
- How to use
- Descriptor Calculator
- Fingerprints Calculator
- Tools
- FAQ
Here is a brief tutorial for using ChemDes.
- This website is designed in accordance with W3C Standards.
- The site core framework is based on Django. So the back-end code is rigorous and standardized, and it is convenient for developers to expand as the logical section and view section are separated very well.
- This site is distinguished form others that are usually based on Java client, which requires users to have a knowledge of programming. Its runtime environment is an additional layer of software with an own set of technical problems and security risks. Ours use the Javascript and jQuery to accomplish some complex and interactive processes instead of Java to avoid problems above.
- The front-end interface uses Bootstrap from Twitter, which offers a good user experience.
1. Calculating 1D/2D molecular descriptors
- Open http://cbdd.csu.edu.cn/chemdes and you will see the homepage of ChemDes. From the introduction you will get to know the major functions of ChemDes.
- Click the
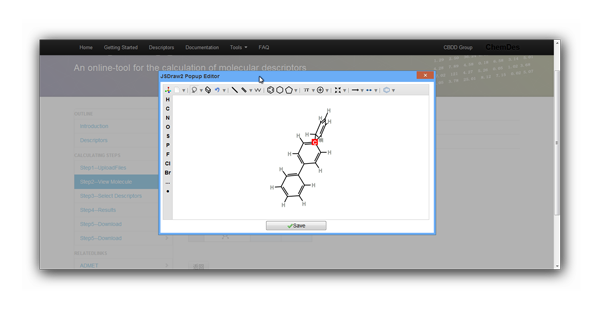
'Web Server'button in the top navigation, and next you will be lead to one certain calculator. - If calculating descriptors of a single molecule, you can use the JSDraw Editor to draw a structure and then click
'Get Smiles/Smarts'button to copy the SMILES to clipboard. Paste the SMILES in the
'Text Box'and choose'view molecular structure'or'submit'to continue.Note: 'view molecular structure' is the function of JSDraw.
If calculating descriptors of a number of molecules at one time, click the
'browse'button and select your file (*.smi or *.sdf) , then click'submit'button.Note:The file should be well checked and prepared before using. You can use 'ChemCONV' or other related software to prepare. The input format must be standard
*.smior*.sdf.-
After the completion of calculation, the browser will redirect users to the result page, In which you will get your result showing and download the result by certain format.
2. Calculating 3D molecular descriptors
The whole process of calculating 3D molecular descriptors is nearly the same with that of Calculating 1D/2D molecular descriptors.
After selecting descriptors, a kind of forcefield for MOPAC optimization is needed and must be chosen from the drop-down menu. For more information please refer ChemMOP.
Note: Limited by the system resources, we set a maximum number of batch computing for each calculator. As the calculation of 3D descriptors includes an optimizing process, the time for calculating should be much longer. Users should wait a bit longer if suspended animation happens.
3. Calculating fingerprint descriptors
refer the process of calculating 1D/2D molecular descriptors
Note: Because of the different definitions of the 42 kinds of fingerprints, the ways to display the results are not the same. The result provides the bits-on and bits-off with 1 and 0.The default is bits-on. At the same time, several kinds of output formats are available for users.
1. Molecular preparation
- We provide different ways to compute descriptors. First, ers just need type in the SMILES, then submit the job. This allows users to calculate one molecule at one time. Second, a
batch computing process can be startup by uploading a file with correct format. (You can get the proper file via ChemCONV)
- Format: The requirement for file format and molecular structure is here:
| Calculator | Supported input format | 3D information |
| Chemopy | *.smi, *.sdf | selectable |
| CDK | *.smi, *.sdf | selectable(contained in the file) |
| RDKit | *.smi, *.sdf | not required |
| Pybel | *.smi, *.sdf | not required |
| BlueDesc | *.smi, *.sdf | required |
| PaDEL | *.smi, *.sdf | selectable |
2. About the speed of calculation
- About the speed of calculation:The main influencing factor is the source packages. Moreover, we have rebuilt some modules related and have optimized the transaction processing systems of server-side. This ensures that the system is stable and possesses considerable computing speed.
3. Batch computing limit
- Batch computing limit : In order to make sure system works properly, and to avoid system suspended animation or session-timeout. We set a limit of the number of molecules uploaded for different calculators. It is listed below:
| Calculator | Count Limit |
| Chemopy | 500 |
| CDK | 500 |
| RDKit | 1000 |
| Pybel | 1000 |
| BlueDesc | 500 |
| PaDEL | 500 |
4. About the result
- We provide output files with different formats according to the different calculators. All of them are listed below.
- If you get other error message or exception, that means the file you upload is not a standard format required. Please check!
| Calculator | Supported output format |
| Chemopy | *.csv |
| CDK | *.csv, *.sdf |
| RDKit | *.csv, *.sdf |
| Pybel | *.csv, *.sdf |
| BlueDesc | *.csv, *.sdf |
| PaDEL | *.csv |
5. Molecular descriptor library
- Molecular descriptor library: Click here for more details.
6. More
1. Molecular preparation
- We also provide different ways to compute molecular fingerprints. First,users just need type in the SMILES, then submit the job. This allows users to calculate one molecule at one time. Second, a
batch computing process can be startup by uploading a file with correct format. (You can get the proper file via ChemCONV)
- Some of the algorithms use geometrical features like geometrical distance matrix. When using the single SMILES to calculate, the system will firstly optimize it with 'mmff94' forcefield. The molecular file uploaded must contain the 3D coordinates.
2. About the result
- About the result:
The calculation result of each type of fingerprint will be given by the
*.csvformat. Four kinds of other formats, namely,'WEKA_HASHED' , 'LIBSVM_MATRIX' , 'LIBSVM_SPARSE','FULL_TAB_UNFOLDED'will be provided when calculating the fingerprints from jCompoundMapper.
Questions from User Feedback
- Why does the error message pop up:Sorry! the program based on RDKit can not identify!The loop will break by this smile,We will show results calculated before this?
- Why does the suspended animation happen when calculating 3D molecular descriptors?
- Why does the blank result page appear when use the file of correct format ?
-
Why does the error message pop up when use the correct
'*sdf'file?
Answers
1. Why does the error message pop up: Sorry! the program based on RDKit can not identify!The loop will break by this smile,We will show results calculated before this?
ERROR MESSAGES:
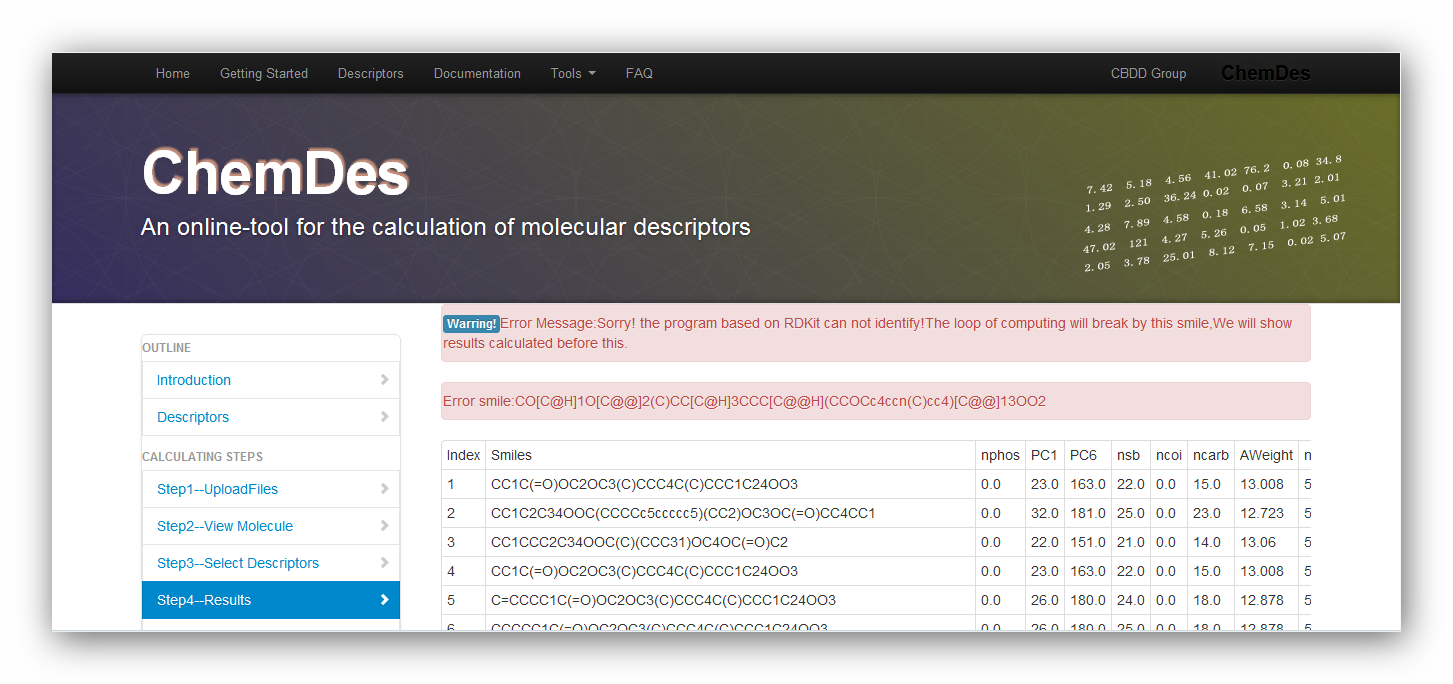

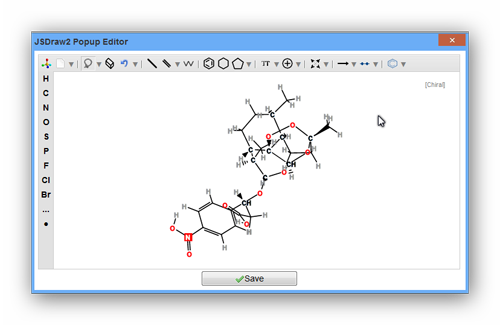
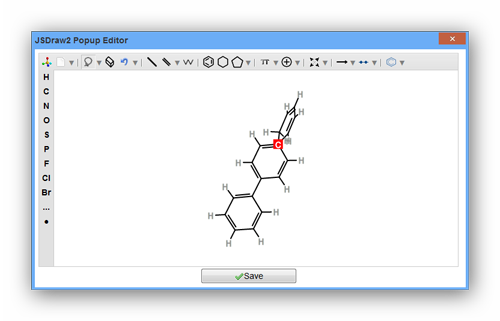
As the pictures above show, the data file is invalid for requirement of RDKit. It may be caused by the reason that one of the smiles is not correct. You can use "view your molecule" function to check your smiles. Or you can drop the one and continue calculating it.
2. Why does the suspended animation happen when calculating 3D molecular descriptors?
It is well known to all that 3D optimization of drug molecules is quite a time-consuming process, such as the pseudo diagonalization, full diagonalization, and density matrix assembling in MOPAC. So we suggest that users should not submit a job containing number of molecules more than 100 when calculating 3D molecular descriptors, especially including someone with large molecular weight.
3. Why does the blank result page appear when using the file of correct format ?
This situation is not commonly encountered. If it happens, please check your file. ave you ever edited the file via other editors that add some special style for the content,for example, colored the string inside? The built-in module requires the plain text without any style.
4. Why does the error message pop up when use the correct'*sdf' file?
It is related with the *.sdf file format.The M CHG and M RAD lines are used for connection tables that ‘overflow’ the allowed atom block limits.By now the M CHG and M RAD lines can not be handled correctly in some cases.